Our proteasome model
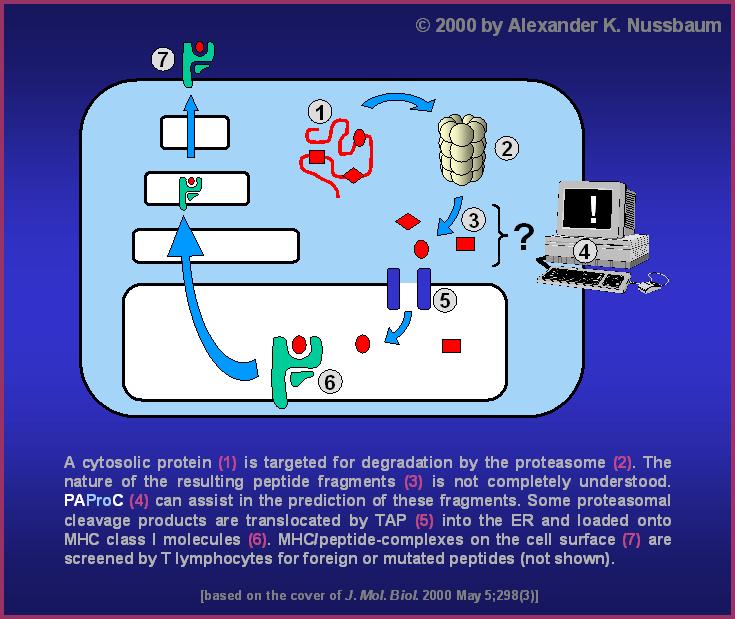
Proteasomes, major proteolytic sites in eukaryotic cells, play an important
part in major histocompatibility class I (MHC I) ligand generation and thus in
the regulation of specific immune responses. Their cleavage specificity is of
outstanding interest for this process.
We constructed computer-based theoretical model proteasomes for the
cleaving of substrate proteins by yeast and human 20S proteasomes.
They were trained by an evolutionary algorithm with the experimental
20S proteasome cleavage data.
The basic assumptions for our model are:
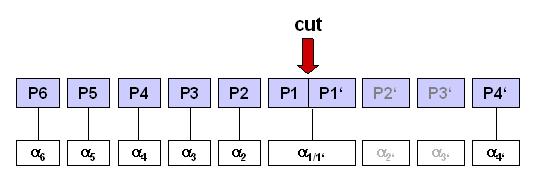
In determing whether to cut or not, the proteasome inspects only a
small neighborhood P6 ... P1 | P1', P4' of the prospective cleavage
site
A main effect results from the affinity of the pair of amino acids in
the P1 and P1' positions to the active subunits in the proteasome. This
effect is modeled by an affinity parameter alpha1(X1, X1').
The value - alpha1(X1, X1') could be interpreted as an affinity of
the pair to the active sites of proteasome.
Each of the positions Pi, i=2,...,6 (or Pi', i=4) exerts
an affinity alphai(Xi) (alphai'(Xi')) towards the prospective
cut which depends on Xi (Xi') but not on the amino acids at the other
positions. The affinities can be positive, negative, or zero.
The model is additive: The total affinity at the position considered is:
The probability of a cut depends only on the total affinity delta. The
cutting process is deterministic: the probability of a cut is equal to 1 if
delta <= 0 and equal to 0 if delta > 0 (mere normalization). This
assumption excludes the occurrence of overlapping fragments.
A stochastic hill-climbing algorithm was used to train the network.
The affinity parameters of the model, which decide for or against cleavage,
correspond with the cleavage motifs determined experimentally.
Proteasome species
Based on different sets of experimental data as learning data, we
received eight different affinity parameter sets ("model proteasomes")
which can be chosen:
Type I: Human erythrocyte proteasome, based on cleavages in enolase
Type II: Human erythrocyte proteasome, based on cleavages in enolase
and ovalbumin peptides
Type III: Human erythrocyte proteasome, based on cleavages in enolase
and other ovalbumin peptides
The yeast proteasomal mutants are denoted by the missing active unit,
all yeast model proteasomes are based on cleavages in enolase.
Sequence name
The sequence name can contain any letters and numbers plus
the characters "_" and "-". Example for
non-permissible characters: spaces, /, (, ), [, ], etc.
The sequence name is only used for your own
information.
If you submit non-permissible characters with your
sequence, you receive the following error
message:
We are sorry, the input (for example the name of the sequence) seems to contain illegal characters.
It may be a problem with ä, ö, ü for example or an empty space within the name of the sequence, which is not allowed for reasons of security. Also most of the special
(i.e. non-alphabetical or non-numerical) characters are not allowed (neither in the name nor in the sequence itself).
If this is not the case, please send an email to christina.kuttler@uni-tuebingen.de.
Submitted amino acid sequence
Amino acid sequences must be entered in the one-letter code. PAProC
will not tolerate additional characters in the sequence, except for
spaces, numbers and newlines, as found in standard sequence formats
such as the FASTA- or original SWISS-PROT-format.
Capital letters or small letters can be used without any
difference. However, the output of the sequence will always be in
capital letters.
If you submit non-permissible characters with your
amino acid sequence, you receive the following error message:
We are sorry, the sequence input seems to contain illegal characters.
Maybe you used a character that does not describe an amino acid ? .
If this is not the case, please send an email to christina.kuttler@uni-tuebingen.de.
Substitute X
The letters "X" and "x" can be used as a "wildcard", in order to
substitute otherwise empty
amino acid positions (for example when your sequence is too short, see
minimum length). Note that "X/x" has a neutral (zero) effect on the
cleavage decision.
Minimum length of submitted sequence
Please note that it only makes sense to test/submit a sequence of at
least 10 amino acids in length. This is to avoid boundary effects in the
model which is based on a window of 6 amino acids to the left
(N-terminal) and 4 amino acids to the right (C-terminal) of a
prospective cleavage site. If you want to test a shorter sequence, you
can fill in extra "X" at both ends of the sequence
(see Substitute X ).
Output style
For the results, you can choose between a) a short output format, b) a
long output format and c) a format only yielding the predicted
cleavage positions as a list of numbers.
a) The short format is very graphical and shows
the cleavages inserted into the submitted sequence as red,
vertical bars. The number of amino acids per line can be chosen in
this format ( short
form example ).
b) The long format will give more detailed information in a
table on cleavage positions and approximate cleavage strength. For more
details check the
example for long
form (use your Browser's back button to return).
c) List of predicted cleavage positions: Numbering starts with the
N-terminus of the submitted sequence as position 1. The list contains
all positions of predicted P1-residues, i.e. amino acid residues
directly N-terminal ("to the left") of the cleavage site. For nomenclature
see the picture
Epitope-destroying cleavages
Important note: PAProC does not predict fragments,
but cleavage sites. The cleavage sites predicted by
PAProC are most likely not all used in the same substrate molecule.
Different combinations of cleavage sites will be used in different
individual substrate molecules, thus producing the typical overlapping
fragment spectrum known from proteasomal digestion experiments
(
example ). It is well-established that the existence of possible
proteasomal cleavage sites within CTL-epitopes does not necessarily
abrogate the presentation of these epitopes on the cell surface.
Therefore, epitope-destroying cleavages predicted by PAProC are not
necessarily executed in each single substrate molecule, thus leaving
enough CTL-epitope intact for presentation by MHC class I.
Interpretation of PAProC results for the
generation of CD8+ T cell epitopes
Most PAProC users are interested in proteasomal generation of CD8+ T cell
epitopes. We would therefore like to give some help for the interpretation
of PAProC predictions assessing the generation of such epitopes.
According to current literature, proteasomes are crucial in the generation
of the correct C-termini of CD8+ T cell epitopes (Craiu et al., 1997;
Stoltze et al., 1998), but not so much for their N-termini, which probably
can be trimmed to the correct size by cytosolic or ER-resident proteases.
Together with what is mentioned above in the paragraph on
epitope-destroying cleavages, we therefore recommend the following rules
for the interpretation of the prediction results by PAProC:
1. Check for the prediction of the correct C-terminal cleavage
(This is by far the most critical criterium for the generation of a CD8+ T
cell epitope.)
2. Check for epitope-destroying cleavages
(This criterium is far less critical. However, when comparing two cases in
which the C-termini are predicted to be generated - e.g. the processing of
epitopes containing point mutations or cleavages of constitutive vs.
immunoproteasome - , this criterium can become decisive.
Last update: 13.4.2005
More detailed information about this program can be found in the
following publications:
C. Kuttler, A.K.
Nussbaum, T.P. Dick, H.-G. Rammensee, H. Schild, K.P. Hadeler, An algorithm
for the prediction of proteasomal cleavages,
J. Mol. Biol. 298 (2000), 417-429 ,
and
A.K. Nussbaum, C. Kuttler, K.P. Hadeler, H.-G. Rammensee, H. Schild, PAProC: A Prediction Algorithm for Proteasomal Cleavages available on the
WWW, Immunogenetics 53 (2001), 87-94
For comprehensive background information, please refer to
From the test tube to the World Wide Web - The cleavage specificity of
the proteasome (A.K. Nussbaum, Dissertation, University of
Tuebingen, Germany, 2001).
The use of PAProC is restricted to non-commercial
purposes.

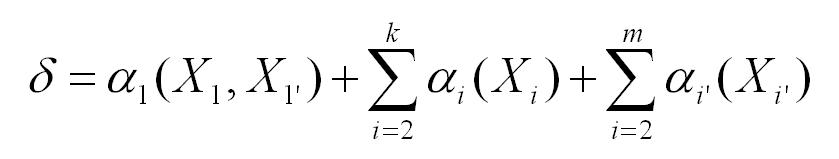
![Inner surface of the yeast 20S proteasome; yellow: inhibitors bound to the three active sites [Groll et al. (1997), Nature].](http://www.paproc2.de/paproc1/20szit.jpg)